
The Reagan-Udall Foundation report on FDA Center for Tobacco Products procedures is now available. It contains recommendations for improving FDA CTP operations, but recognizes that two of the serious impediments that the CTP faces are the huge number of applications for new products and aggressive litigation by the industry.

Several recommendations deal with improving transparency and streamlining processes to make reviews more systematic and predictable. Doing so will also increase FDA efficiency and help industry know what to expect when submitting applications for new products.
A key R-U recommendation to accomplish this goal appears on page 19 of their report:
Consider clarifying in formal policy the Center’s plans for triaging its substantive reviews to conserve resources when there are certain critical sections of the application that can be indicative of whether all sections of the application merit review.
Right now Premarket Tobacco Product Applications (PMTAs) have to include a wide range of information, ranging from product toxicity to effects on tobacco consumption behavior and product switching. This leads to long complex applications that require a lot of work by applicants to prepare and for the FDA to assess. For e-cigarettes at least, this process can be streamlined by beginning with known effects of e-cigarettes in general, then asking whether a company proposing a specific new product presents evidence that the proposed product will have different effects than e-cigarettes in on average.
This is precisely the approach that FDA appropriately applied when concluding that Logic menthol e-cigarettes were not “appropriate for the protection of public health,” the legal standard for authorizing new tobacco products. In addition to continuing the apply this approach to other flavored e-cigarettes, including menthol, FDA should apply this approach to other key aspects of e-cigarettes.
Specifically, FDA should make initial triage decisions starting by asking if a proposed e-cigarette behaves differently from e-cigarettes in general in <<XX>> ways:
On average, adult smokers who use e-cigarettes as consumer tobacco products are no more likely to stop smoking cigarettes than adult smokers who do not use e-cigarettes
The foundational assumption that e-cigarettes are an effective product for harm reduction is that smokers will “switch completely” from cigarettes to e-cigarettes. In contrast, actual real-world evidence shows no such benefit for e-cigarettes overall. In particular, several meta-analyses, including our meta-analysis (below), consistently show that adult smokers who use e-cigarettes as consumer tobacco products are no more likely to stop smoking cigarettes than smokers who do not use e-cigarettes. (Newer long-term longitudinal studies show depressed quitting among e-cigarette users.) The first triage decision is whether a specific product behaves differently from the average.
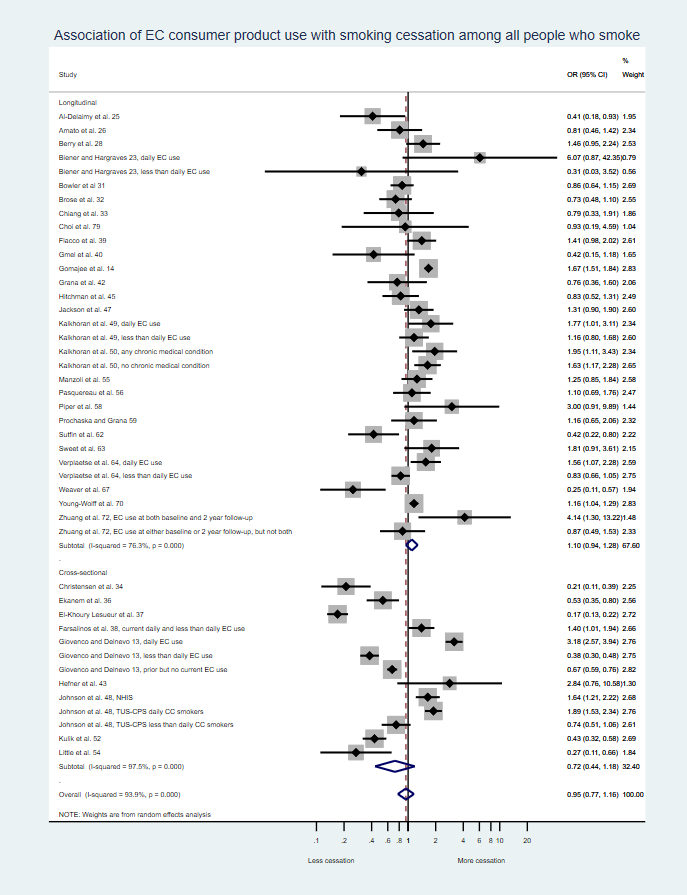
We did find that daily e-cigarette users quit smoking more than non-users, but non-daily e-cigarette users quit less. But, unlike prescription medicines, CTP does not have the authority to specify how e-cigarettes are used. The thing that matters for determining whether e-cigarettes are APPH depends on the overall use in the whole population.
This distinction between prescription medicines and consumer products is very important. In contrast to observational studies of e-cigs as consumer tobacco products summarized above, there are also randomized clinical trials showing that e-cigs have efficacy for cessation when used as a therapy under medical supervision. The RCT results would be relevant if an e-cig company submitted its product for approval as a medicine by the FDA Center for Drug Evaluation and Research (CDER). CDER can limit where and how products are sold and has much broader authority over how products are used such as by specifying use frequency and limiting use to by prescription. The therapeutic vs. consumer product distinction is important because CDER and CTP have fundamentally different missions and standards.
In contrast to RCTs, observational studies assess the effects of e-cigarettes as consumer products on cigarette smoking is what is relevant to the Center for Tobacco Products assessment of e-cigarettes as consumer tobacco products and they show that, on average, e-cigarettes as consumer tobacco products are not associated with increased adult smoking cessation.
An important detail in assessing whether a specific product leads smokers to “switch completely.” The definition FDA accepted when authorizing Philip Morris’ IQOS accepted Philip Morris’ definition that 70% was switching completely even though that meant that 30% of users remained dual users, which is likely still enough cigarette smoking to have substantial disease risks. FDA should define switch completely rigorously as meaning stopping smoking cigarettes among virtually all smokers (say 90-95% of smokers). What FDA means by “switching completely” should universally apply to all PMTAs, and should not be determined by each applicant on a case-by-case basis.
Thus, just as it has done when assessing menthol in specific e-cigarettes, FDA should leave the door open to applicant companies demonstrating that their specific e-cigarette, as actually used, leads to complete switching.
E-cigarettes on average have broad appeal to youth, surpassing conventional cigarettes and other tobacco products
In addition to effects on adult smokers, to find that e-cigs are APPH, FDA must determine their effects on youth non-users, as well as adult non-users. Unlike prescription medicines, e-cigarettes, like other consumer products, are widely available to and used by youth, despite so-called youth-access measures.
FDA accepts the fact that there is widespread e-cigarette use by youth and uses it in making decisions about specific e-cigarette products; it is the basis for not authorizing the sale of flavored e-cigarettes and, so far, one menthol e-cigarette. Again, this fact provides the context for assessing specific products. If an applicant could demonstrate that their product performs differently from e-cigarettes on average, appeal to youth would no longer be a reason to triage an application and making a negative APPH decision.

On average, youth and young adults who begin tobacco use with e-cigarettes are 2-4 times more likely to start smoking cigarettes than youth and young adults who do not use e-cigarettes
The third triage question is whether an applicant presented convincing evidence that their specific product did not have this effect (summarized below).

In particular, FDA should require that applicants demonstrate that specific product characteristics and marketing would be aversive to youth and other never-tobacco users. Assurances that the company did not intend the product to be used by youth or was applying youth access controls would not be adequate since there are decades of experience that such statements are disingenuous and that access controls are weak. (Indeed, some of the lawsuits against Juul show how the company manipulated its product design and youth access controls to limit effectiveness [link1, link2].)
Any marketing restrictions designed to deter youth should be specified by FDA in its Marketing Granted Order and made a requirement of marketing, and should not be left to be determined in post-market review after the damage had been done to youth.
Making triage decisions beginning with these three facts implements FDA’s current operational definition of APPH
These three facts speak directly to the FDA Center for Tobacco Products’ two key considerations about whether a specific proposed product is “appropriate for the protection of public health,” the standard in the law:
(A) the increased or decreased likelihood that existing users of tobacco products will stop using such products; and
(B) the increased or decreased likelihood that those who do not use tobacco products will start using such products.
The first fact speaks to item (A): If smokers do not switch completely from cigarettes to e-cigarettes, i.e., become dual users or relapse back to sole cigarette smoking, they will not benefit from any theoretical reduced harm associated with the fact that e-cigarettes deliver lower levels of some toxins than cigarettes.
The second two facts speak to item (B): If e-cigarettes expand the nicotine market to include youth who were not already smoking there is harm because, regardless of any benefits of e-cigarettes compared to cigarettes these considerations are moot, since it is uncontested that using e-cigarettes is more dangerous than not using e-cigarettes among never smokers. In addition, if initiating nicotine use with e-cigarettes leads users to add cigarettes (i.e., become dual users), they are even worse off.
Moving from the average to a specific product
As discussed above, FDA is required to make decisions about authorizing the marketing of specific products not broad categories of products on average.
FDA can, however, use these general patterns to assess specific product applications by beginning their assessment of individual product PMTA applications by asking the question: Has the applicant presented compelling evidence that their product is sufficiently different from the average that
- smokers and other tobacco product users will switch completely (as rigorously defined by FDA) to the proposed e-cigarette, and
- the proposed product has characteristics than are different enough from the average that it will not appeal to youth, young adults and others who have never used nicotine or to former smokers.
If the answer to both questions is “yes,” FDA CTP would then proceed to a more detailed assessment, including toxicology.
Otherwise, the application would be denied because it does not meet the threshold standard for appropriate for the protection of public health, thereby avoiding all the work of considering the many other factors that would have to be assessed for specific products that meet the threshold standard.
As noted above, this is the precise approach FDA appropriately applied when determining that Logic menthol e-cigarettes were not appropriate for the protection of public health. The current approach to flavors, including menthol, should become part of the triage process recommended by the Regan-Udall Foundation.
Conclusion
If a specific proposed product PMTA does not pass these critical threshold criteria, there would be no need for FDA to undertake the time-consuming process of assessing the specific toxicity of the product or other issues that FDA would assess in a full application.
This approach could allow creation of a two-step process where applicants began with applications limited to these three areas without going to the expense of toxicological and clinical health studies. If a specific product passed triage, the FDA could invite the applicant to submit these additional data as well as other materials currently requested in a PMTA. Doing so would reduce work and speed up the process, which, as Regan-Udall noted, is in everyone’s interest.
(This post is an updated version of an earlier blog post on how to streamline evaluation of new products by starting with key questions about use.)
