
On February 16, 2021, Mitch Zeller, Director of the FDA’s Center for Tobacco Products (CTP), published an update on FDA’s progress in handling the waterfall of applications for premarket authorization of new tobacco products (PMTA) and other applications from tobacco companies.
Everyone was expecting a large number of PMTA applications for new products to come in by the September 9, 2021 deadline, but the volume is hard to conceive. While FDA was still reviewing applications, they had already processed applications for 4.8 million products from 230 companies and accepted applications for 84,000 products. (“Acceptance” of the application does not mean the FDA has authorized sale of the product, just that all the basic elements of the application are there.) Of the 84,000 accepted, FDA has filed 29,000, which means that they are ready for substantive review.
FDA is selecting filed applications to process at random, but “due to the large number of ENDS products currently marketed and for which we anticipated receiving submissions, FDA decided to dedicate a portion of its resources to reviewing the products that account for most of the current market.” This is a fair and sensible procedure.
Even with this prioritization, however, the FDA is facing a gigantic task in assessing all these applications, particularly because a full review of an accepted PMTA application includes “full reports of all information concerning investigations which have been made to show the health risks of the tobacco product and whether the tobacco product presents less risk than others; full statements of the components, ingredients, additives, and properties, and of the principle or principles of operation; and full descriptions of the methods used in, and the facilities and controls used for, the manufacture, processing, and, when relevant, packing and installation of, the tobacco product.”
FDA could streamline this process by beginning with three well-established facts
- On average, adult smokers who use e-cigarettes as consumer tobacco products are no more likely to stop smoking cigarettes than adult smokers who do not use e-cigarettes. (Additional information)
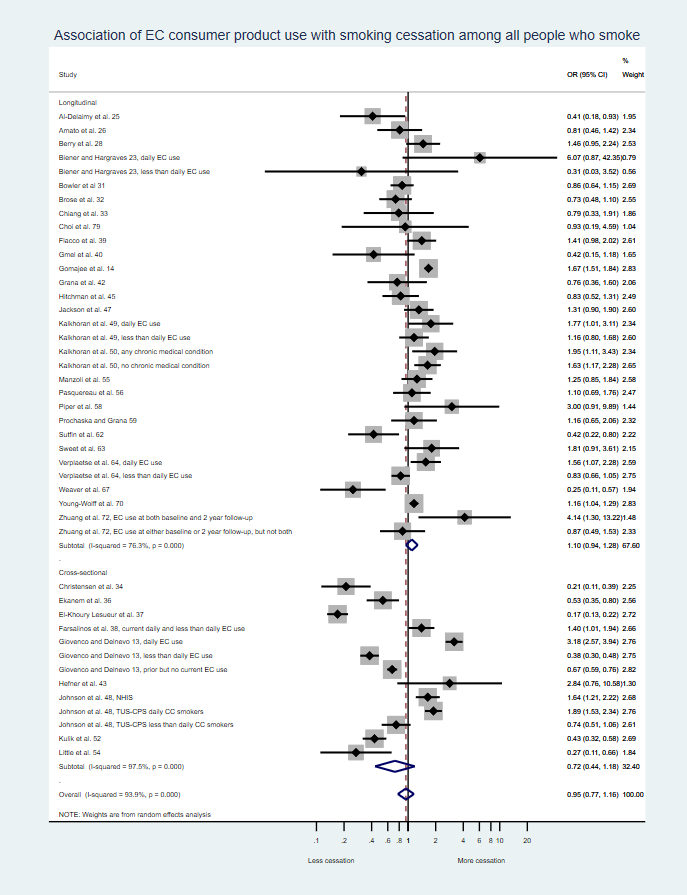
In contrast to these observational studies of e-cigs as consumer tobacco products, there are also randomized clinical trials showing that e-cigs have efficacy for cessation when used as a therapy under medical supervision. The RCT results would be relevant if an e-cig company submitted its product for approval as a medicine by the FDA Center for Drug Evaluation and Research (CDER). That process is separate and distinct from the Center for Tobacco Products’ assessment of PMTA applications being discussed here.
The therapeutic vs. consumer product distinction is important because CDER and CTP have fundamentally different missions and standards. CDER can limit where and how products are sold and has much broader authority over how products are used such as by specifying use frequency and limiting use to by prescription. CTP does not have any such authority to specify conditions of use or use patterns required for efficacy.
But these observational studies assess the effects of e-cigarettes as consumer products on cigarette smoking is what is relevant to the Center for Tobacco Products assessment of e-cigarettes as consumer tobacco products and they show that, on average, e-cigarettes as consumer tobacco products are not associated with increased smoking cessation.
- E-cigarettes on average have broad appeal to youth, surpassing conventional cigarettes and other tobacco products.

- On average, youth and young adults who begin tobacco use with e-cigarettes are 2-4 times more likely to start smoking cigarettes than youth and young adults who do not use e-cigarettes.

Specifically, these three facts speak directly to the FDA Center for Tobacco Products’ two key considerations about whether a proposed product is “appropriate for the protection of public health,” the standard in the law:
(A) the increased or decreased likelihood that existing users of tobacco products will stop using such products; and
(B) the increased or decreased likelihood that those who do not use tobacco products will start using such products.
The first fact speaks to item (A): If smokers do not switch completely from cigarettes to e-cigarettes, i.e., become dual users, they will not benefit from any theoretical reduced harm associated with the fact that e-cigarettes deliver lower levels of some toxins than cigarettes.
The second two facts speak to item (B): If e-cigarettes expand the nicotine market to include people who were not already smoking there is harm since, regardless of any benefits of e-cigarettes compared to cigarettes are moot, since it is uncontested that using e-cigarettes is more dangerous than not using e-cigarettes among never smokers. In addition, if initiating nicotine use with e-cigarettes leads users to add cigarettes (i.e., become dual users), they are even worse off.
Moving from the average to a specific product
FDA is, however, charged with making decisions about authorizing the marketing of specific products not broad categories of products on average.
FDA can, however, use these general patterns to assess specific product applications by beginning their assessment of individual product PMTA applications by asking the question: Has the applicant presented compelling evidence that their product is sufficiently different from the average that
- smokers and other tobacco product users will switch completely to the proposed e-cigarette, and
- the proposed product has characteristics than are different enough from the average that it will not appeal to youth, young adults and others who have never used nicotine.
If the answer to both questions is “yes,” FDA CTP would then proceed to a more detailed assessment.
Otherwise, the application would be denied because it does not meet the threshold standard for appropriate for the protection of public health, thereby avoiding all the work of considering the many other factors that would have to be assessed for specific products that meat the threshold standard.
In making these decisions, FDA should define switch completely vigorously as meaning stopping smoking cigarettes among virtually all smokers (say 90-95% of smokers). The definition FDA accepted when authorizing Philip Morris’ IQOS accepted Philip Morris’ definition that 70% was switching completely even though that meant that 30% of users remained dual users.
Applying this approach to Juul
Based on its own publicly reported data, Juul does not come close to meeting this standard:
- Juul’s study Switching Away from Combustible Cigarettes across a 12-Month Period Among Adult Smokers who Purchased the JUUL® System found that at one year 51.2% of smokers who bought a device directly from Juul switched completely to Juul.
- Another Juul study, Longitudinal Study of Dual Use of Combustible Cigarettes and the Juul® System in Adult Current Smokers at 12 Months, found that 53.1% switched and that among those who did not switch 89% (41.6%/46.9%) were dual users.
Without questioning the findings, these two studies showed about half the users did not switch completely and most of the rest went from being smokers to being dual users. These two studies by themselves show that Juul fails the first criterion.
With regard to the second criterion, FDA should also require that applicants demonstrate that specific product characteristics and marketing would be aversive to youth and other never-tobacco users. Assurances that the company did not intend the product to be used by youth or was applying youth access controls would not be adequate since there are decades of experience that such statements are disingenuous and that access controls are weak. (Indeed, some of the lawsuits against Juul show how the company manipulated its product design and youth access controls to limit effectiveness (link1, link2)
Conclusion
Answering these questions will be relatively easy for the products currently on the market because the FDA will have the benefit of past experience when making these decisions, particularly how the proposed product differs from the product on the market.
If a proposed product PMTA – as exemplified by Juul — does not pass these both these two threshold questions, there would be no need for FDA to assess the specific toxicity of the product or other issues that FDA would assess in a full application.
Doing so would reduce work and speed up the process, which is in everyone’s interest.
